
Geoffrey A. Charters MSc (Hons) PhD (Pathology)
Molecular oncopathologist
Geoffrey A. Charters MSc (Hons) PhD (Pathology)
Molecular oncopathologist
Biography
PhD research
For a panel of human metastatic melanoma cell lines (NZM), to:
For the NZM cell line panel, to:
Geoffrey commenced work on his PhD in February 1998 at the Auckland Cancer Society Research Centre (ACSRC) under the supervision of Prof. Bruce Baguley, Dr Andrew Shelling, and, before his move to the Department of Molecular Medicine, Dr Graeme Finlay. The project proposal envisaged a continuation of his Masters work with G1 cell cycle regulation in human metastatic melanoma cell lines by investigating the retinoblastoma-associated protein mediated checkpoint as this was believed to be functionally defective in all melanomas (see "Background" tab). The source material was to be a panel of melanoma cell lines being developed at the ACSRC, the NZMs.
One branch of the research would have a functional basis, and would seek to determine if the pRB subsystem was operative in the cell lines. This would be achieved by challenging the cells with cultural conditions that were expected to result in G1 arrest only if the pRB system were intact, and the denial of serum growth factors was selected as the most tractable approach. Control and serum-deprived cultures would be harvested at intervals and the distribution of cells throughout the cell cycle determined by flow cytometry. If the hypothesis under test were correct, and the pRB subsystem was functionally defective in all melanomas, none should arrest in G1.
A second branch would investigate the genomic integrity of the chromosome 9p21 region. This is a frequent site of disruptions in melanoma that may affect genes for two proteins that regulate pRB function, p15 and p16. Here, deletion mapping based on the attempted amplification with PCR of a set of markers in the region with well-established ordering would be the technique employed. The genetic integrity of these genes, CDKN2A and CDKN2B would also be explored, initially using the single-strand conformation polymorphism analysis, and ultimately with DNA sequencing. The gene for pRB itself, RB1, was to be explored, but this was expected to be a challenging exercise as it is a complex gene with 27 exons spanning over 200 kbp. Here, an attempt was to be made to use reverse-transcriptase PCR to generate cDNA for analysis.
A third branch would seek to determine if the key proteins under investigation, pRB, p15, and p16 were actually expressed. Here, the options of using Western blotting and flow cytometry existed. Since antibodies for use in flow cytometry must be validated using Western analysis first in order to prove their specificity, Western analysis was the method of first choice.
Data obtained during his Masters research had shown that at least one of the NZMs, NZM7, existed in a state of heteroploidy, complicating cell cycle analysis. This type of analysis was to be central to the PhD project, so an early part of the project was to be the development of robust assays for the accurate flow cytometric determination of ploidy and detection of heteroploidy.
It is in the nature of original research that some of the investigations intended will go well and some poorly. New thoughts on how to address the key questions can give rise to extensions to parts of the study, and interim results may yield surprises that open unanticipated avenues for research. There is therefore a tendency for these projects to evolve, and the destination and length of the journey are not always those envisaged at the start. Such was the case here.
Of the work intended that went well, excellent methods were developed for the extraction and stringent quantitation and standardisation of DNA and protein, and for the robust determination of absolute ploidy. The test of pRB function proved enlightening, although it involved quite large scale experimentation, with 960 5 mL cultures being grown to assay the entire NZM panel. PCR-SSCP analysis proved to be a very effective mutation screening tool.
Of the things that went badly, perhaps the chief disappointment was the failure to develop a means of producing RB1 cDNA despite many months of effort, the issue being thought to be a high G:C component in exon 1. Lacking this, genetic analysis of RB1 was abandoned as impracticable due to the time that would have been required to develop PCR-SSCP methods to screen the 27 exons.
Western blotting, never an easy technique, proved quite difficult to optimise for all of the targets attempted. This was particularly true for pRB where mediocre results were necessarily accepted until a very late stage of the research when redevelopment based on a different antibody ultimately permitted an outstanding technical quality to be achieved.
Among the extensions to the planned work were the investigation of the alternate first exon of CDKN2A and the expression of the ARF protein which it encodes. While this is not directly involved in the pRB subsystem, being encoded by the same gene as p16, a rare if not unique phenomenon in eukaryotic genomes, there was debate in the literature as to which encoded protein was the functional target of deletion or mutation in melanoma.
The studies of pRB expression had originally been intended to establish presence or absence only, with no regard for phosphorylation status. The inclusion of phosphatase inhibitors during the redevelopment of the pRB Western blotting methodology that took place late in the project allowed some data to be gathered concerning this, with a surprising result.
Even though a formal loss of heterozygosity study was not warranted for the 9p21 region given the lack of matched normal DNA, this was undertaken and did produce data that validated and clarified other findings, and, incidentally, provided genotype information that proved very useful for cell line discrimination.
The most significant extension to the intended work came about principally as a result of the analysis of ploidy. The heteroploidy that had previously been seen in NZM7 was seen in other cell lines also, suggesting that it may be a common feature of melanoma. More interestingly, individual strains of NZM7 that had been produced by single cell propagation in order to provide uniploid subclones for experimentation were again found to be heteroploid after a short time in culture. This established that the affected cell lines were not merely heterogeneous, but that they were genomically unstable. This was recognised as a potentially important finding as if it correctly reflected the in vivo situation, it could provide a mechanism both for the tumorigenesis of melanoma and for its notorious resistance to therapy.
This raised the obvious question of the cause of this instability and a thorough investigation of the literature led to the conclusion that centrosomal numerical dysregulation was almost certainly the basis for this. The project was therefore extended to seek an answer as to whether this was in fact occurring, and immunocytochemistry and fluorescence microscopy were the chosen tools.
.jpg)
NZM2 cell cycle phasing
In response to serum deprivation some cell lines arrested in G1 despite having a flaw in the pRB system, in this case, the deletion of CDKN2A and CDKN2B.
Red = G0-G1; green = S; blue = G2-M. Closed symbols = control; open symbols = serum deprived at day zero.
Figure 1

p16
Expression of p16 was negligible in all but the NZM6 and NZM7 (highlighted) cell lines and its derivatives.

NZM3 pRB

NZM7 pRB
Unphosphorylated pRB increased in response to prolonged time in culture and serum deprivation in most cell lines (e.g. NZM3), but in NZM6, NZM7 and its derivatives, no phosphorylated pRB was observed, even in actively proliferating control cultures.
A and B represent experimental replicates.
Figure 3
Geoffrey found that some of the NZM cell lines arrested in G1 immediately upon deprivation of serum growth factors, as would be expected only if the pRB subsystem were intact (Fig 1). This observation was amenable to two interpretations, either that the pRB subsystem was indeed intact in some cases, disproving the primary hypothesis under test; or alternatively, that it was not the sole arbiter of G1 progression as was believed. In either case, the finding was novel. It also was the first hint that the aberrations of the pRB subsystem that are frequently observed in cancer might not be as a result of their causing a loss of proliferative control, but for some other reason.
Unexpectedly, evidence was found that in some of the cell lines where the G1 arrest mechanism was flawed, deprivation of serum growth factors caused cells to accumulate in G2 or mitosis, these states not being distinguishable by the flow cytometric method used. While it was known that inhibitory growth factors can cause arrest in G2 this was the first evidence that some factors may be essential for passage through it. The study did not extend to the point of determining if the accumulation was due to an absolute barrier to progression, or merely a large increase in transit time, nor to seeking to isolate or identify the factor responsible.
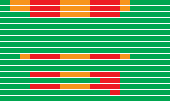
Deletion map
Each bar depicts the status of each of the 17 markers probed in the 9p12 - 9p23 region of an NZM cell line. CDKN2A is to the right.
Green = present; orange = microsatellite absent; red = exon absent.
Not to scale.
Figure 2

D9S974
Two cell lines derived from NZM7 by single cell propagation differ in genotype, as shown in this comparison of allele lengths for the 9p21 microsatellite marker D9S974, situated between the CDKN2A and CDKN2B genes.
Figure 4
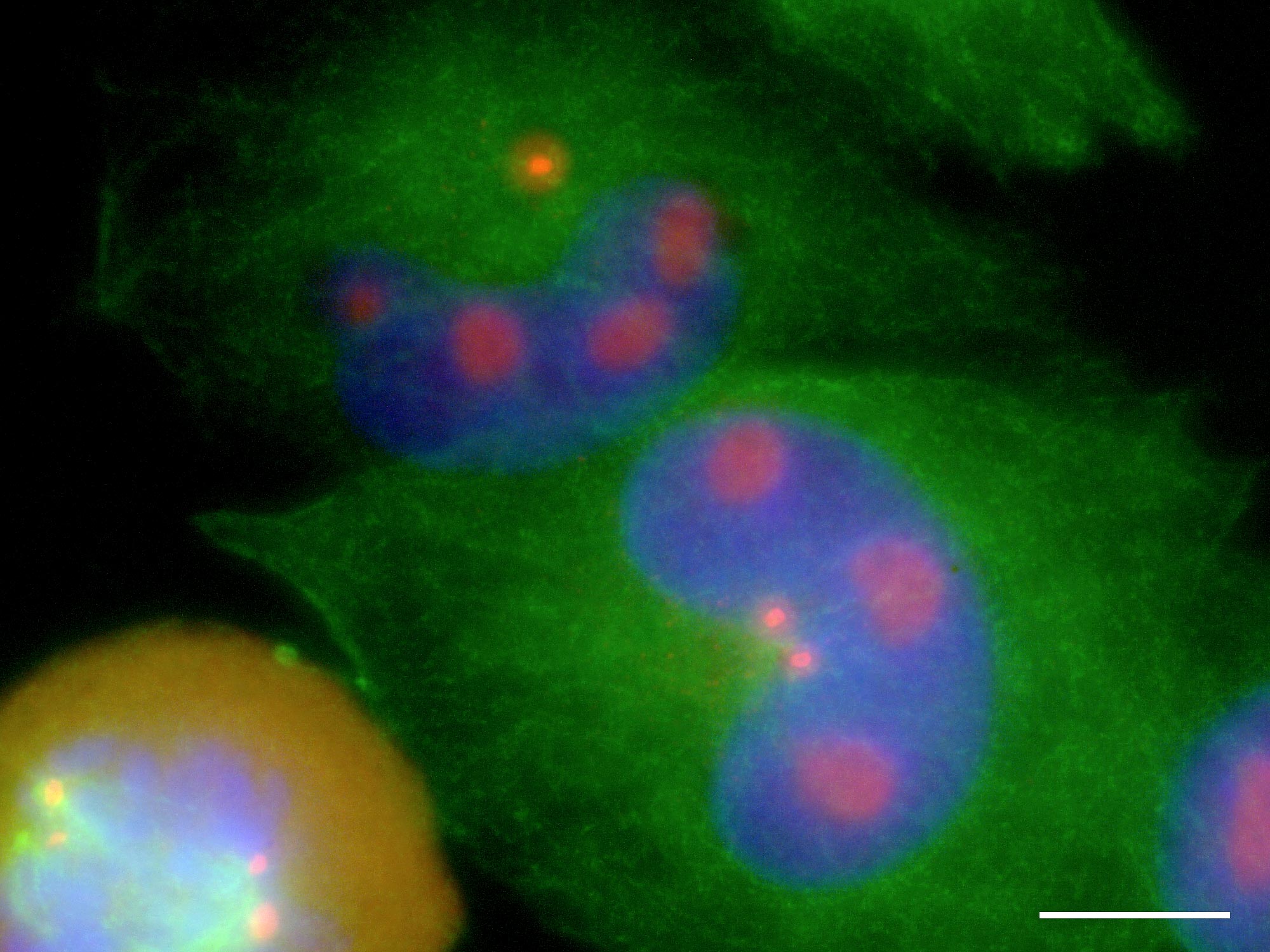
NZM9
Pericentrin accumulates in nucleolar reservoirs during interphase and floods the cytoplasm upon nuclear envelope breakdown.
Green = tubulin; blue = DNA; red = pericentrin. Scale bar = 10 µm.
Figure 6
Deletion analysis revealed the loss of some part of the 9p21 locus in about half of the cell lines. In all of these, CDKN2A was affected, and in all but two CDKN2B was also affected (Fig 2). Two polymorphisms were found, but neither had significance for the encoded protein. Interestingly however, in one of these cases the NZM7.2 cell line was heterozygous, while the related NZM7.4 cell line retained only one variant. These two cell lines were derived by single cell propagation from the parental NZM7 line, itself heterozygous for the polymorphism. The inference from this was that the NZM7.4 cell line had suffered a loss of heterozygosity event. This may have occurred before or after its independent establishment, but in either case it argued for genomic instability in NZM7. This was not surprising since it was because of the finding of heteroploidy in NZM7 that separate lines were established by single cell propagation in the first place. Of significance was the fact that several of the lines where CDKN2A was deleted still arrested in G1 in response to serum deprivation. This was further evidence that the loss of p16 in melanoma was not necessarily because this abrogated pRB proliferative control.
Western analysis eventually established that little if any p15 was being expressed by those cell lines where its gene was intact, but it could be dramatically induced by treatment of the NZM4 cell line with TGF-β.
Little p16 expression was seen, even among those cell lines where the gene was intact, but large quantities were observed in NZM6, NZM7, and the two subclones of NZM7 studied, NZM7.2 and NZM7.4. This proved to be an important observation as when robust pRB expression data were obtained it was found that while all of the NZMs expressed apparently normal sized pRB, in these cell lines expressing abundant p16, pRB existed only in the hypophosphorylated, growth-suppressive state (Fig 3), as might be expected if p16 were operating normally to prevent the phosphorylation of pRB by CDK4; however, the extracts involved were from cells that were proliferating vigorously.
The only examples of proliferation in the presence of unphosphorylated pRB to be found in the literature were associated with cells in which both RAS and MYC were aberrantly active. Most significantly, a colleague investigating BRAF in these cell lines had found that they contained an activating mutation. The current hypothesis is therefore that BRAF activation is functionally equivalent to RAS mutation, and the cell lines also contain activated MYC. This hypothesis is yet to be explored.
Observations from several lines of enquiry converged to suggest that human metastatic melanoma cell lines were heterogeneous in nature, in particular the heteroploidy observed in several cell lines and the difference in CDKN2A genotype seen between NZM7.2 and NZM7.4. It should be mentioned that microsatellite analysis results made if very unlikely that NZM7.4 was not a genuine descendant of NZM7, indeed, a microsatellite marker adjacent to CDKN2A that was also heterozygous in NZM7 and NZM7.2 was hemizygous in NZM7.4 (Fig 4). The return to heteroploidy of these uniploid isolates demonstrated that this instability was on-going and led to an investigation of centrosomal regulation by immunocytochemistry and fluorescence and confocal microscopy.
This study proved to be extremely fruitful. In all of the cell lines studied, a significant number of mitotic cells with abnormal centrosome numbers were found, and there was a great deal of evidence to support the conclusion that these resulted in malmitoses with aberrant genome partitioning (Fig 5, below). The direct consequence of this is a failure to maintain euploidy, and with that, the ongoing generation of genomic and thence phenotypic diversity. Given the possible significance this had for tumorigenesis and therapeutic evasion in melanoma, this was a finding of considerable potential significance, and one not previously reported.
This immunocytochemical study also provided some interesting incidental data, chief among which was the observation that pericentrin, the centrosomal marker being used, accumulated in the nucleolus of cells during interphase, and on nuclear envelope breakdown was released into the cytoplasm (Fig 6). Since this is transported to the centrosomes and its presence there is a prerequisite for microtubule nucleation, this may be a mechanism by which the initiation of mitotic spindle formation is synchronised with the completion of prophase, or more fundamentally, with the completion of S-phase and the duplicated genome being ready for partitioning. This was a novel observation that gave rise to a surprising inference: in several published studies of pericentrin, none involving melanoma, any such nucleolar reservoirs should have been detected and reported, yet they were not. The implication is that they were absent, and it may be that this behaviour is a characteristic of melanocytic cells alone, or possibly, an aberration only seen in melanoma, in which case it may represent a therapeutic opportunity. This is yet to be explored.
The serum deprivation results established that in some cell lines where CDKN2A was deleted, G1 arrest still occurred suggesting that pRB regulation of proliferation was intact despite the absence of p16. If loss of p16 is not sufficient to ensure loss of proliferative control, then what might be the role that p16 plays in normal melanocytes, which if not performed, conduces them to become melanoma? In a fascinating development, it was recently reported that p16 has an important function in centrosomal regulation, and the observation here that centrosomal dysregulation is widespread in melanoma cell lines is unlikely to be coincidence. It is entirely possible that in the molecular aetiology of melanoma, the loss of genomic stability is a more fundamentally important factor than the loss of proliferative control. This need not be surprising since where genomic instability exists, and there is rapid generation of genetic and phenotypic diversity, it will only be a matter of time before proliferative control is lost in a variant that arises. The question of why loss of p16 favours the development of melanoma in particular is as yet unresolved.
Geoffrey's thesis was accepted by the University of Auckland in December 2007, and he received his doctorate from the Chancellor at the Autumn Graduation Ceremony at the Auckland Town Hall on May 9, 2008.
The path to his doctorate had been a long and challenging one, and he attributes his eventual success to two things above all else: first, his steadfast determination to persevere no matter how long it took, what obstacles needed to be overcome, or whatever the emotional or financial price was that he had to pay; and second, the constant and unflinching support of his supervisors, Prof. Bruce Baguley, Assoc. Prof. Andrew Shelling, and Dr Graeme Finlay, and that of the Department of Molecular Medicine and Pathology and the Cancer Society of New Zealand, to all of whom he is forever indebted.


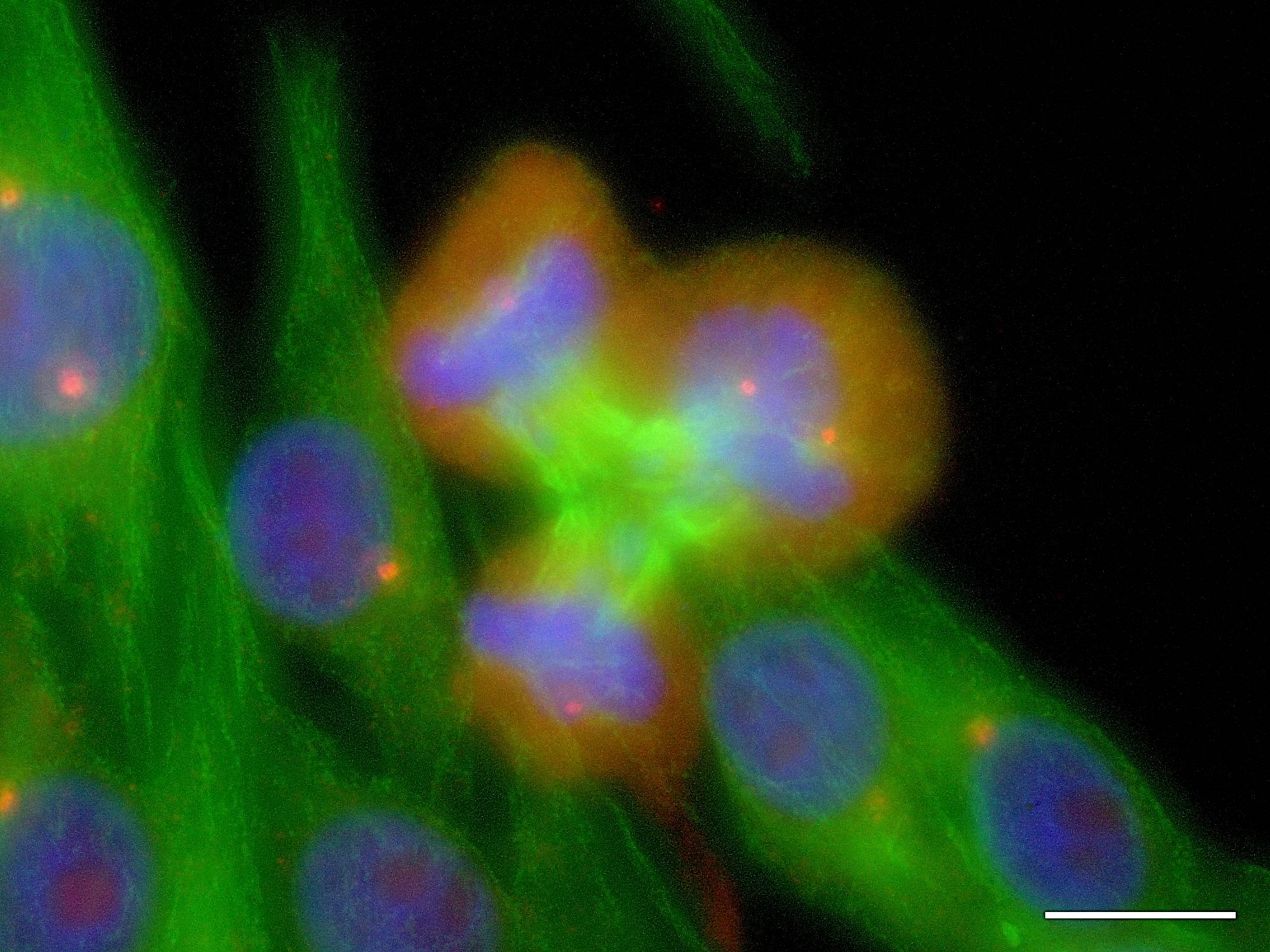
Ternary mitoses in NZM12
In these fluorescence microscopy images of melanoma cells tubulin, an element of the cytoskeleton and mitotic spindle apparatus, is shown in green, DNA and chromatin in blue, and pericentrin, a centrosome-associated protein in red. Centrosomes, being foci of tubulin nucleation, generally appear as orange points. Scale bars = 10 μm.
These images depict NZM12 cells undergoing ternary mitoses under the influence of supernumerary centrosomes: metaphase at left, anaphase in the centre, and telophase at right. It is fundamentally impossible for every cell arising from such a division to be genetically identical to the parent cell.
These images constitute irrefutable evidence that centrosome numerical dysregulation occurs in metastatic melanoma cell lines, and that ongoing genetic, and thence phenotypic diversity will result. If cells in melanoma tumours behave similarly then this dysregulation may well be contributing significantly both to the development of melanoma itself, and to its notorious resistance to existing therapies.
Figure 5